La variación genética natural en el genoma garantiza la biodiversidad e impulsa la evolución. Sin embargo, dado que los procesos evolutivos naturales requieren milenios, no podemos esperar a que adapten los cultivos a las condiciones climáticas rápidamente cambiantes.
por la Universidad Heinrich-Heine de Düsseldorf

Para salvaguardar la seguridad alimentaria mundial, los investigadores deben acelerar la identificación de variantes naturales de ADN adecuadas para mejorar el rendimiento de los cultivos en condiciones de estrés.
Un equipo de investigación dirigido por el Dr. Thomas Hartwig y la Dra. Julia Engelhorn, del Instituto de Fisiología Molecular de la HHU y el MPIPZ, presenta un nuevo y eficiente método para mapear los «interruptores» genéticos de las plantas en una publicación reciente en Nature Genetics . Estas pequeñas secciones del genoma, que no son genes en sí mismas, determinan cuándo, dónde y en qué medida un gen está activo. Son comparables a un regulador de intensidad que regula la intensidad de una lámpara.
Si bien hasta ahora la investigación se ha centrado principalmente en los genes en sí, el nuevo estudio demuestra que las diferencias clave entre plantas (por ejemplo, la variación de tamaño o la resistencia a enfermedades o situaciones de estrés) a menudo no están determinadas por los genes, sino por estos interruptores reguladores. Sin embargo, tradicionalmente no solo es difícil localizar estas regiones con precisión, sino también determinar qué cambios son decisivos. Esto está cambiando gracias a un nuevo método de mapeo escalable desarrollado en el marco del proyecto.
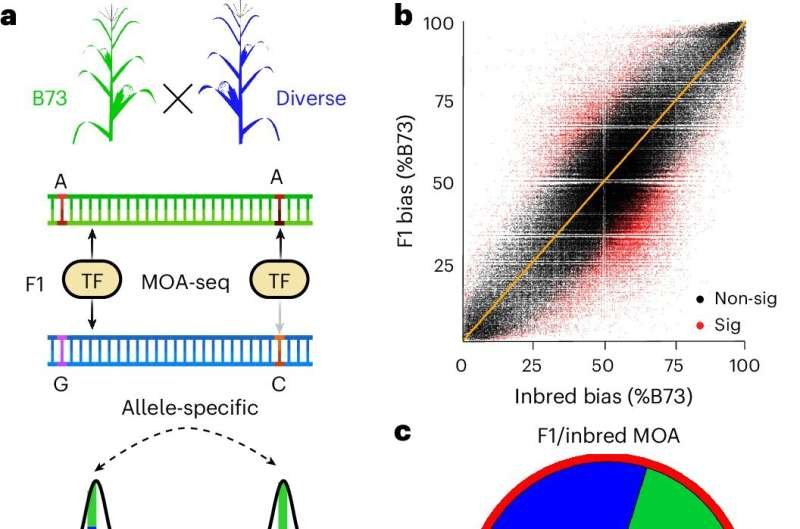
El equipo de investigación analizó 25 híbridos de maíz diferentes, es decir, cruces de distintas variedades de maíz, identificando más de 200.000 regiones en el genoma donde las variaciones naturales influyen en los interruptores reguladores.
Engelhorn, autor principal del estudio, afirma: «Aunque estos interruptores reguladores representan menos del 1% del genoma, las variaciones a menudo explican una parte sustancial de las diferencias en los rasgos hereditarios, a veces más de la mitad».
Hartwig, autor correspondiente del estudio, comenta: «Comprender cómo funcionan estos interruptores reguladores proporciona una nueva y poderosa herramienta para mejorar tanto la resiliencia como el rendimiento de los cultivos, sentando las bases para procesos de mejoramiento más inteligentes en el futuro».
Nature Genetics (2025). DOI: 10.1038/s41588-025-02246-7
Los investigadores aplicaron su método específicamente a los rasgos que juegan un papel en el estrés por sequía, identificando más de 3.500 interruptores reguladores individuales y los genes asociados a través de los cuales las plantas responden a condiciones limitadas de agua.
Engelhorn afirma: «Nuestro enfoque permite la comparación directa de las diferencias en las variantes de cambio heredadas por vía materna y paterna en un solo experimento. De este modo, podemos ofrecer a la comunidad investigadora del maíz un recurso de más de 3500 sitios reguladores vinculados a la sequía, lo que abre nuevas posibilidades para ajustar la expresión génica y lograr una mayor robustez».
Hartwig añade: «La precisión de este mapeo nos permite aprender de las diferencias naturales en los interruptores y cómo funcionan, lo que a su vez permite una manipulación específica de los interruptores para desarrollar plantas con características mejoradas».
Esta investigación se realizó en colaboración con un equipo de la Universidad de California en Davis, del que forma parte la Dra. Samantha Snodgrass. La coautora del estudio destaca el cambio de perspectiva que conlleva este enfoque: «A pesar de décadas de investigación exitosa, gran parte del genoma —las partes externas a los genes— sigue siendo una caja negra. Este nuevo método revela nuevas posibilidades y nos permite identificar la función de estas áreas no codificantes, proporcionando a biólogos y criadores nuevos objetivos precisos para nuevos enfoques de investigación y desarrollo».
El estudio se llevó a cabo dentro del Clúster de Excelencia CEPLAS en Ciencias Vegetales en HHU y MPIPZ.
Más información: Julia Engelhorn et al., La variación genética en los sitios de unión de factores de transcripción explica en gran medida la heredabilidad fenotípica del maíz, Nature Genetics (2025). DOI: 10.1038/s41588-025-02246-7
